
We are computational scientists who focus on chemical biology, the interactions between small molecules and biological macromolecules. We develop and apply new methods that may be helpful for structure-based drug design. The group is led by David Minh, an associate professor in chemistry.
One of our major efforts is to understand structural mechanisms of activation and to calculate the strength of signaling through seven transmembrane receptors (7TMRs), traditionally known as G protein coupled receptors (GPCRs), and other signaling proteins. We developed a method that combines physics-based molecular simulation with machine learning to identify intracellular pocket conformations and to compute signaling efficacy with an error of less than 20%. Based on this technology, some group members have started a company, Biagon Inc.
Another of our major research areas is implicit ligand theory (ILT), a theoretical framework for binding free energies which David derived in 2012. Most binding free energy calculations involve computationally expensive molecular simulations of flexible binding partners. David showed that, in theory, equally good results may be achieved by computing free energies between flexible ligands and multiple rigid receptor configurations.
Some of our achievements are described below.
Spectroscopic methods have shown that ligands perturb the equilibria of 7TMR intracellular pocket conformations, but the identity of these conformations has remained unclear. We have developed a method that combines molecular simulation and machine learning to identify conformations and their signaling efficacies.
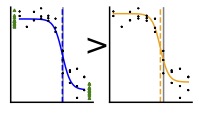
The standard protocol for analysis of concentration response curves involves normalization by the control data but not fitting to it. We showed that including control data in the fit improves the accuracy and precision of parameter estimates.
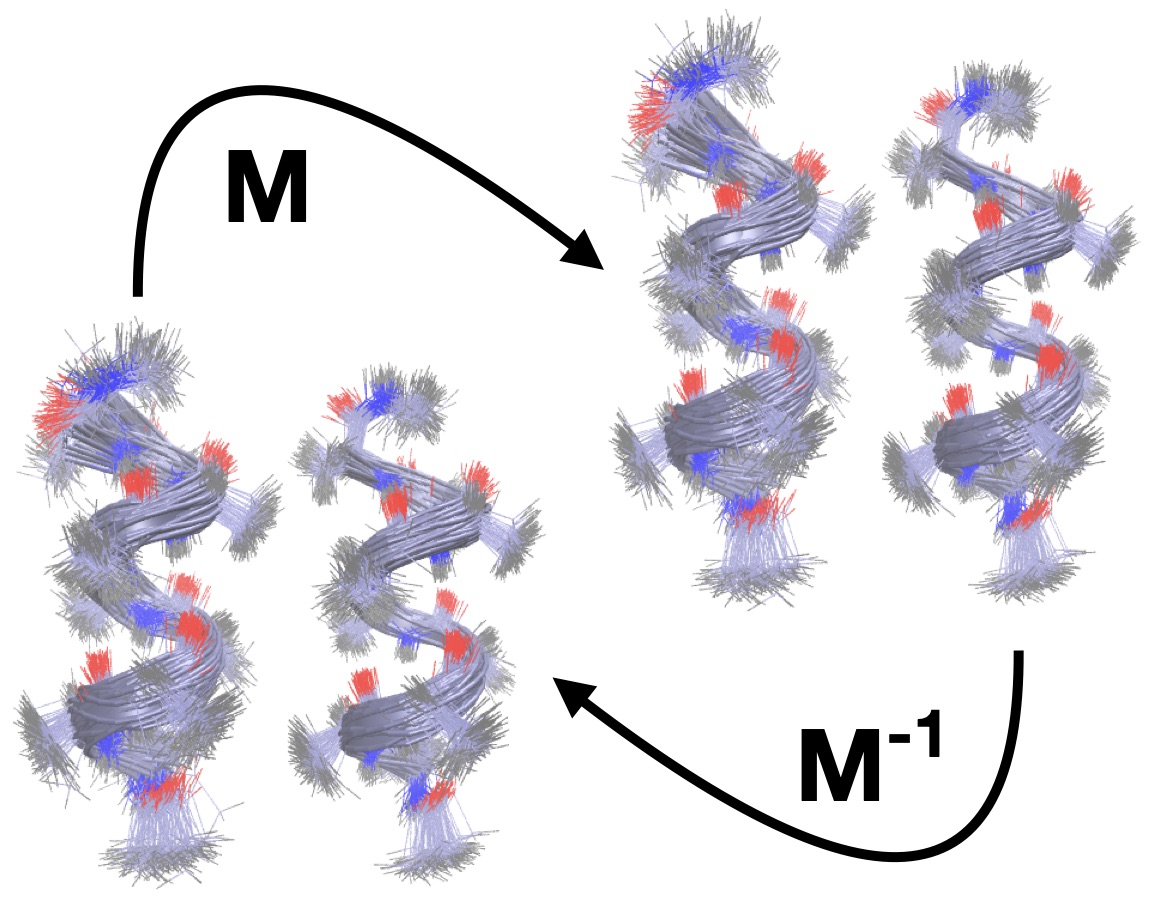
We trained deep neural networks to map between peptide conformations. The learned mappings were used to estimate free energy differences.
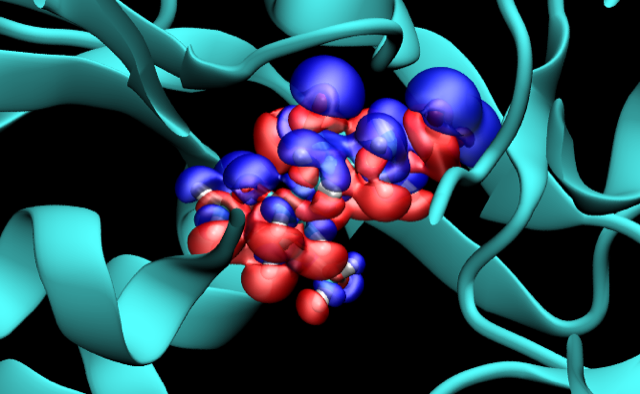
Although it is well-known that ligands are polarized by proteins, the magnitude of this effect had not been quantified in many systems. We evaluated the ligand polarization energy for several hundred protein-ligand complexes and showed that it is a large and highly variable component of the binding energy.
The binding potential of mean force (BPMF), the binding free energy between a flexible ligand and a rigid receptor, is a critical ingredient for esimating binding free energies with implicit ligand theory. We have written software to precisely estimate this quantity and tested it on a diverse set of 85 protein-ligand complexes.
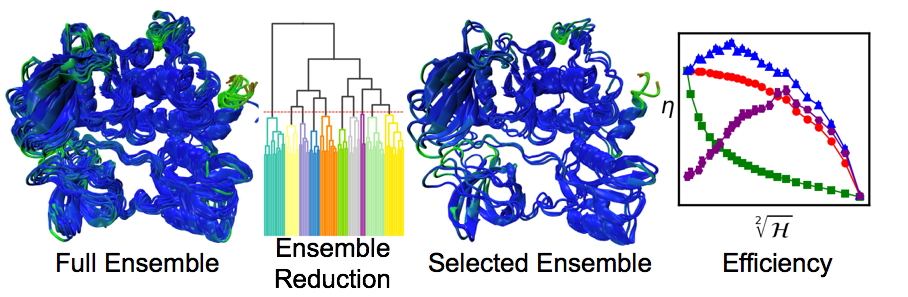
Although there are many ways to select representative snapshots from a molecular dynamics simulation to perform molecular docking, it has been unclear how to assess these methods. We pointed out that this procedure is an example of a statistical method, stratified sampling, and that the efficiency of stratification can be used to assess ensemble reduction methods.
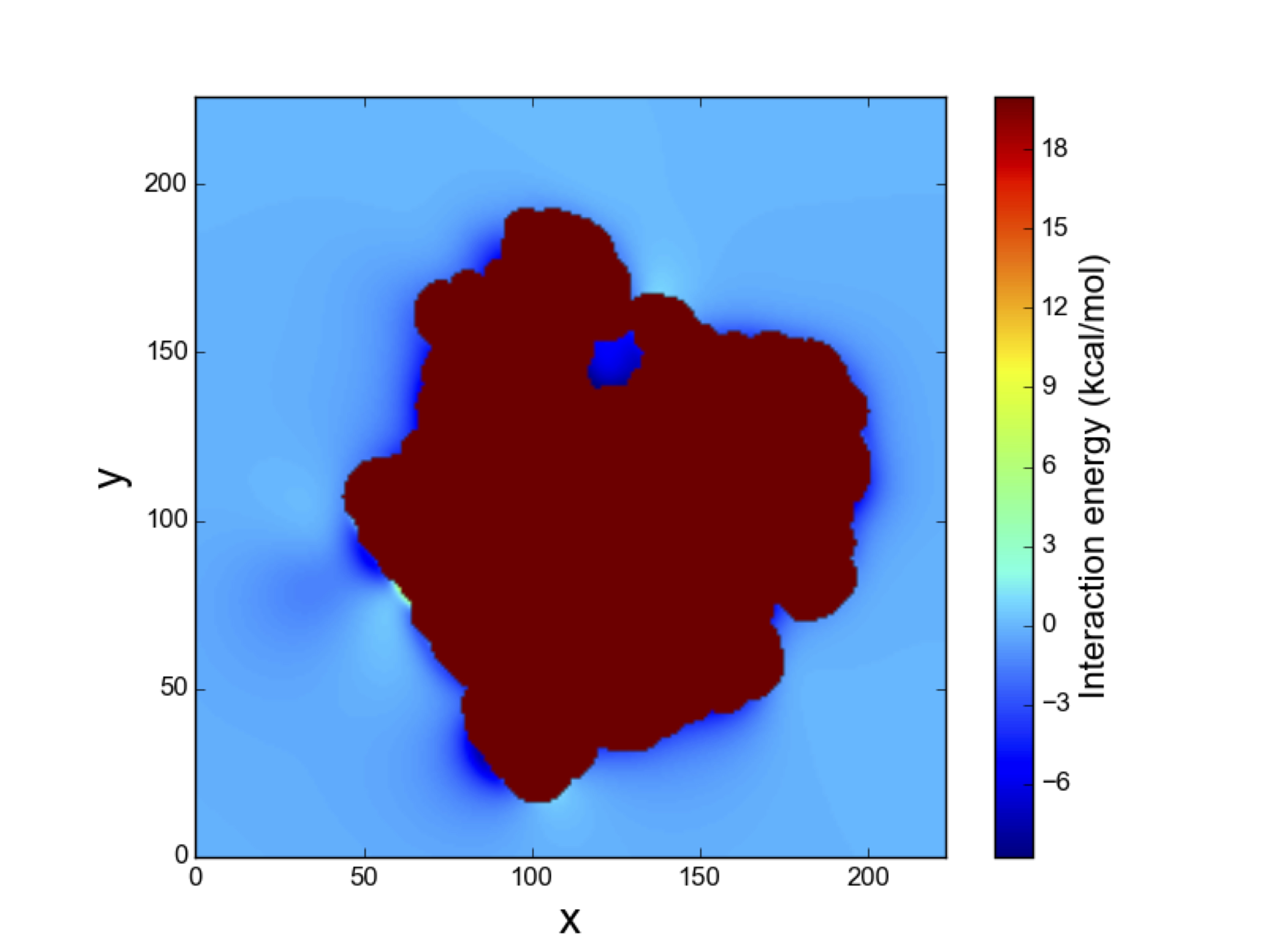
We have shown that the Fast Fourier transform can be used to calculate binding free energies using implicit ligand theory. Previously, the use of FFT in molecular docking was only to estimate interaction energies.
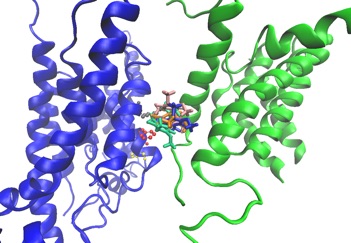
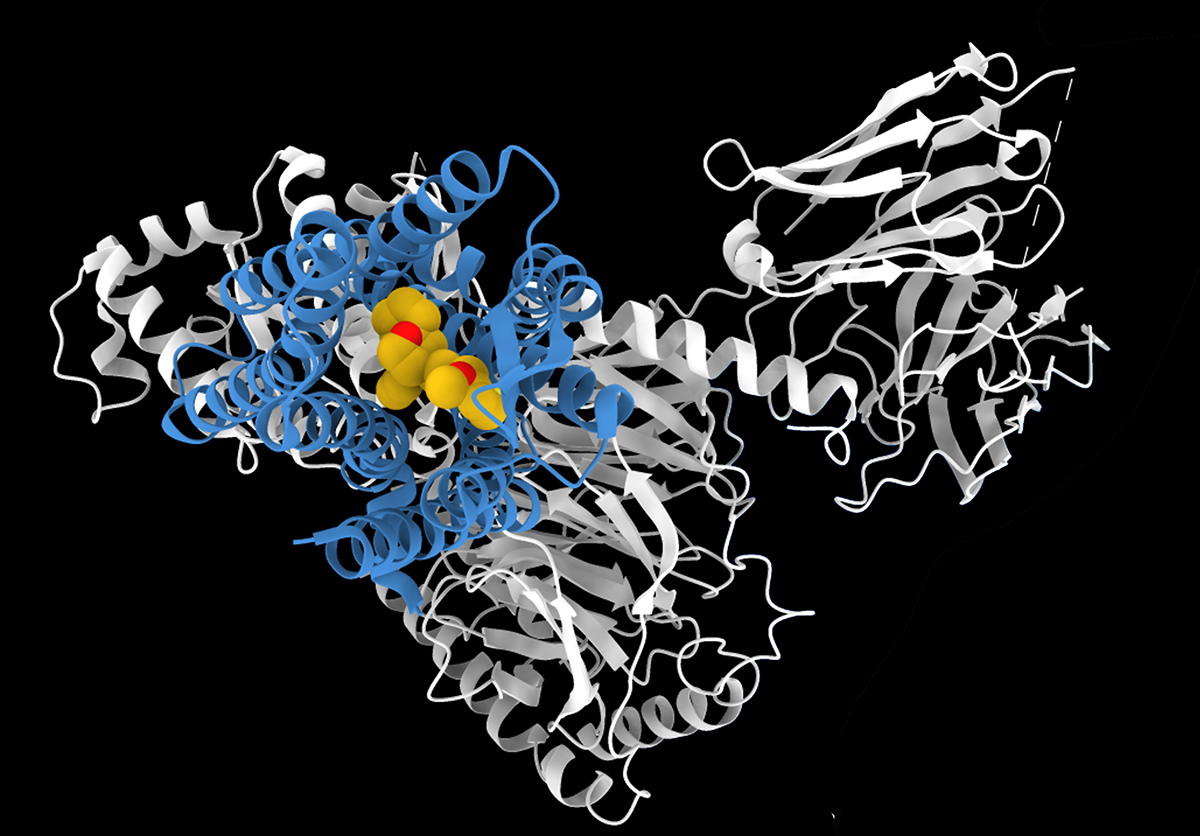
We have identified the ubiquinone binding site in the bacterial ion pump NQR. This binding site is not obvious from the crystal structure. It is a possible target for structure-based drug design.
See article in IIT Today.
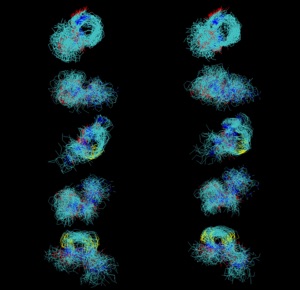
We have shown how to use constrained molecular dynamics, such as torsional dynamics, as a Monte Carlo move for molecular simulation. Previously, molecular simulations based on constrained dynamics would not sample from the appropriate distribution or not sample the entirely of configuration space.
See article in IIT Today.
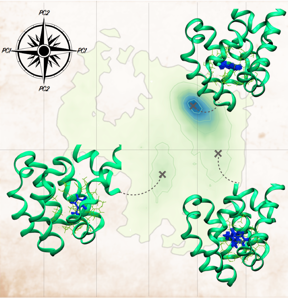
We have performed large-scale protein-ligand binding free energy calculations using implicit ligand theory. Previously, implicit ligand theory was only applied to host-guest systems.
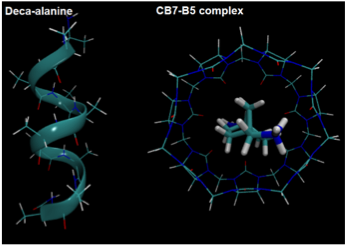
We have shown that if there are a sufficient number of states in a replica exchange simulation, the precise definition of states does not affect sampling efficiency. Previously, many scientists thought that the number of states in replica exchange should be carefully optimized.
This website contains information about: our research projects in signaling protein activation, fast binding free energy calculations, enhanced sampling methods, data analysis methods, and modeling metabolic enzymes from pathogenic bacteria; a complete list of our publications from the lab and David’s prior work; links to source code and data related to publications and classes; links to recommended software; a nascent scientific blog; some information about our members and alumni; some group photos; and finally information about visiting or joining the lab.
Our research has been supported by


Robert E. Frey, Jr.
David gave the keynote presentation Metaphors in Protein Motion at the conference Metaphors of Life: From Science to Text, hosted online by "Ion Ionescu de la Brad" Iași University of Life Sciences, Romania.
May 16, 2026Congratulations to Liliia for graduating with her B.S.! We wish her the best of luck in her Ph.D. program.
May 12, 2026David chaired a session and presented ML Mapping of GPCR Intracellular Pocket Conformations to Multidimensional Pharmacological Efficacy at the Biophysics of Membrane Reactions in the Brain conference in Bucharest, Romania.
April 16, 2026Congratulations, Joseph, on your successful Ph.D. thesis defense!
April 13, 2026Congratulations, Ella, on your successful Ph.D. thesis defense!
November 15, 2025David was invited to present Computational Molecular Pharmacology at the Center for Biocatalysis and Bioprocessing Annual Conference at the University of Iowa (Coralville, IA).
September 1, 2025Welcome to our RES-MATCH students Fatih and Fatimah!
August 12, 2025Congratulations to Talha for winning the Welcome Week Student Poster competition (Graduate)!
May 30, 2025David presented AI: Drugs that send the right chemical messages at ShapeShift.
May 20, 2025David gave an invited talk, ML Mapping from GPCR Conformations to Multidimensional Pharmacology, at the 4th GPCRs-Targeted Drug Discovery Summit in Cambridge, MA.
April 4, 2025David gave a contributed talk, Machine learning maps intracellular pocket conformations to multidimensional signaling efficacy, at the Midwest AI for Drug Discovery and Development Workshop at the University of Chicago.
March 17, 2025David presented Computational Molecular Pharmacology in the Department of Pharmacology Seminar Series at Northwestern University Feinberg School of Medicine.
February 26, 2025Congratulations, Cookie, on your successful Ph.D. thesis defense!
December 2, 2024David was invited to present Computational Molecular Pharmacology at RTI International.
November 26, 2024David presented the seminar Computational Molecular Pharmacology at the Duke University Department of Chemistry, where he met with Nobel laureate Robert Lefkowitz.
September 16, 2024Welcome to our new AI & ML engineer Vigneshwaran!
September 4, 2024Welcome to our new student Michal!
August 14, 2024Congratulations to David for winning the Welcome Week Student Poster competition (Undergraduate)!
August 14, 2024Biagon Inc. featured in an Illinois Tech News Article and the IIT Research magazine.
July 25, 2024David presented Bayesian Regression Links Biochemical and Cellular Efficacy of the Coronavirus Main Protease, work by Van La (Sophie) in collaboration with Lulu Kang, at PyData Chicago.
June 24, 2024Welcome to our new student Talha!
May 16, 2024David presented Linking Biochemical and Cellular Efficacy of the Coronavirus Main Protease at the ASAP (AI-driven Structure-enabled Antiviral Platform) Open Science Forum.
April 25, 2024Congratulations to our Chemistry department awardees Ella (Kilpatrick Fellowship), David (Senior Undergraduate Award), and Joseph (Teaching Assistant)!
April 19, 2024Congratulations to the Biagon team, including David, Joseph, and Jim, for winning second prize in the Kaplan Pitch Tank!
April 16, 2024Congratulations on your Ph.D. thesis defense, Sophie!
January 2024Welcome new students Aiman and Brandon!
December 18, 2023David presented Learned Mappings for Targeted Free Energy Perturbation between Peptide Conformations at the 125th Statistical Mechanics Conference at Rutgers University (New Brunswick, NJ).
November 13, 2023David gave a seminar at the University of Illinois Chicago (host: Ao Ma).
Fall 2023Welcome new students Anna, Beomjong, Luis, Poshan, Pranami, Urvi, and Ryan!
May 17, 2023David presented about enzyme kinetics of the MERS coronavirus main protease to the ASAP (AI-driven Structure-enabled Antiviral Platform) consortium.
